关于我们
关于我们





















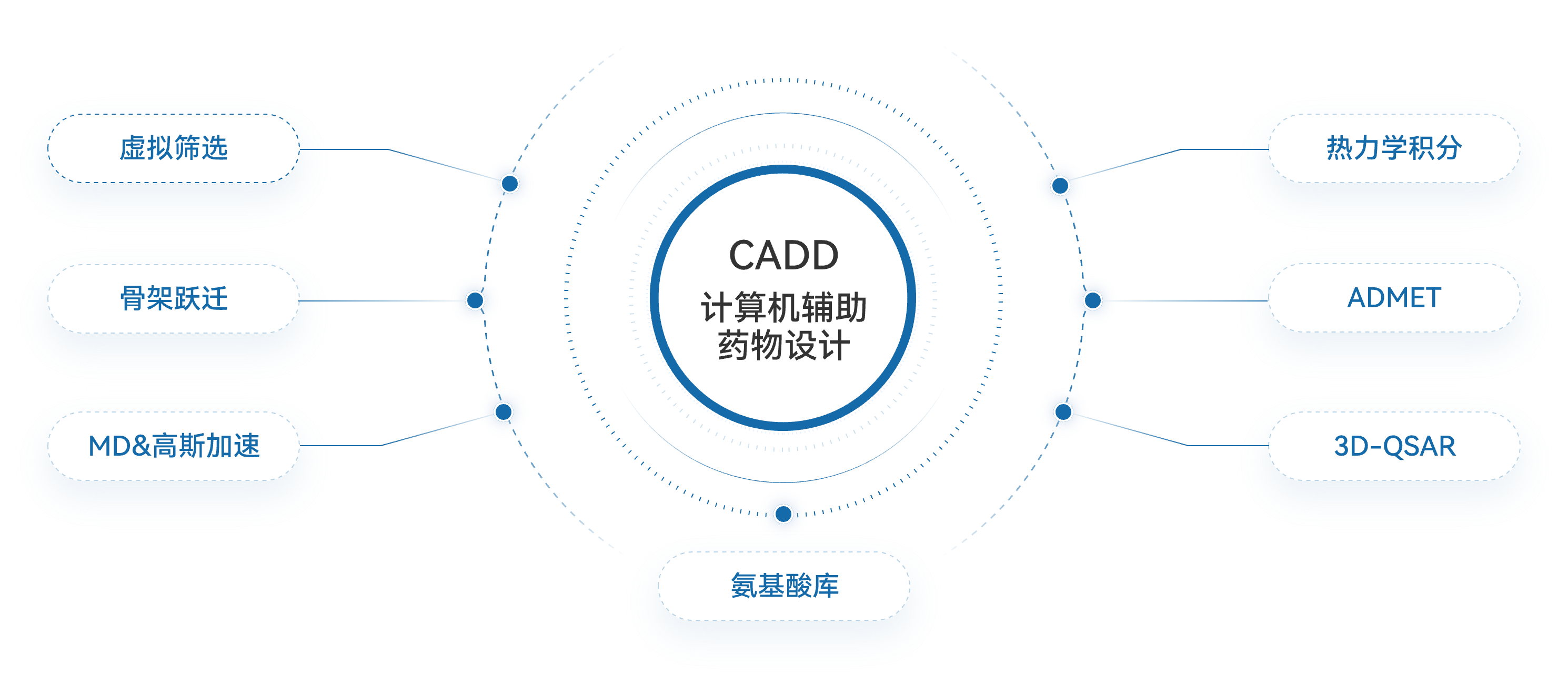
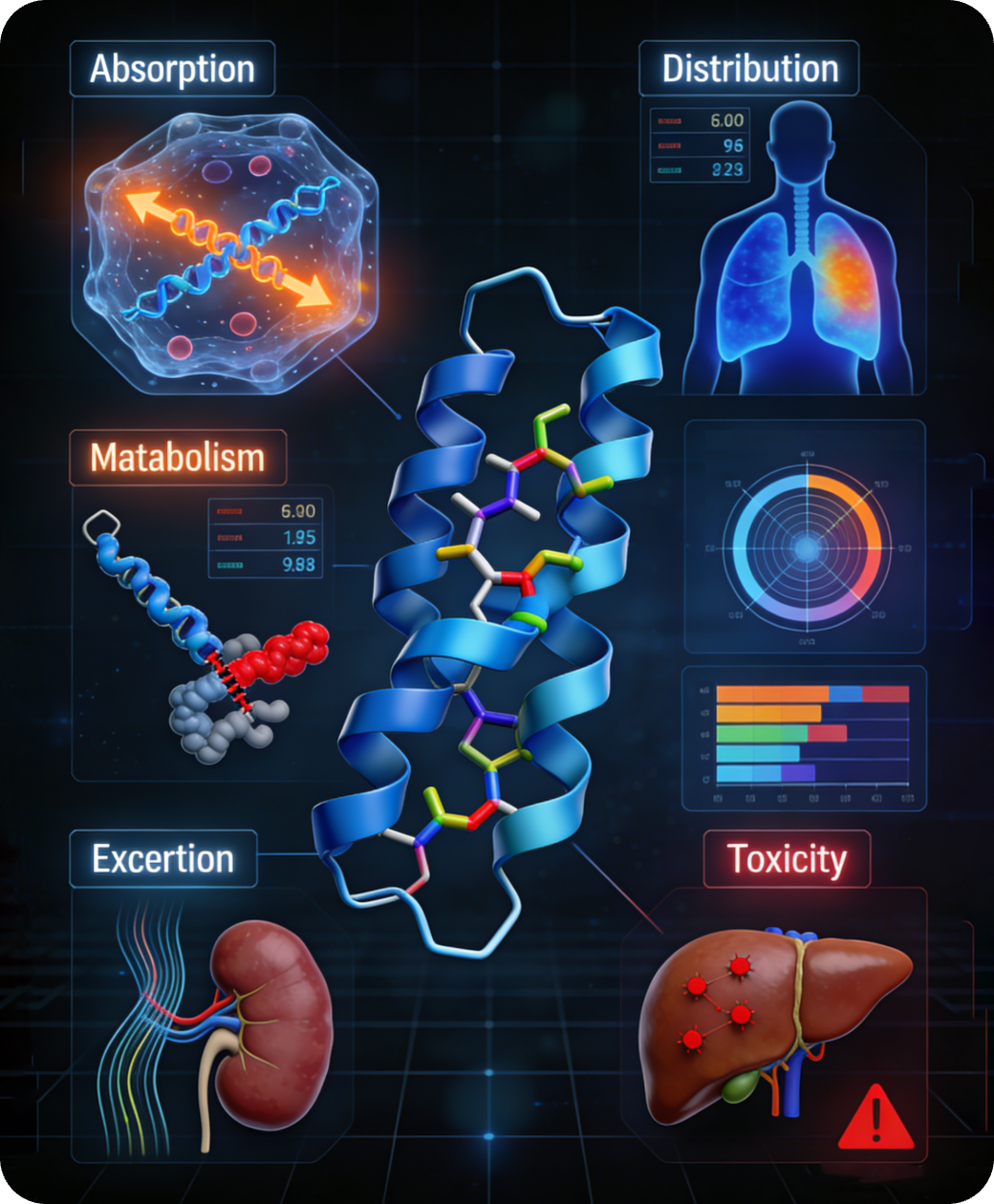
iCVETide®计算化学平台涵盖内容包括虚拟筛选,骨架跃迁,ADMET,氨基酸库等等
诺和晟泰自主研发的iCVETide®计算化学平台,是我们攻坚多肽新药的核心引擎与创新壁垒。平台深度融合计算化学(CADD)与人工智能(AIDD),完整覆盖从靶点早期验证、智能虚拟筛选、先导化合物优化到临床前研究的全流程。
iCVETide®是我们实现多肽药物跨越式研发的“加速器”与“决策中心”。 它驱动着我们同步推进两类战略:一方面,以前沿的AI模拟与设计能力,攻克全新靶点,挑战全球首创(First-in-Class);另一方面,对热门靶点进行极致优化,高效产出具有“同类最优”(Best-in-Class)潜力的候选分子。这一平台能力确保我们能快速布局并推进具备快速上市(First-to-Market)前景的IND项目,系统性突破多肽药物研发的效率瓶颈。
简而言之,iCVETide®代表了我们将计算智能转化为差异化管线优势的核心能力。